Introduction Montmorillonites are one of the more abundant smectitic clays in nature. They are characterized by the substitution of Al3+ in the octahedral sheet by Mg2+, resulting in a negative layer charge that is compensated by hydrated cations adsorbed in the interlamellar space. These cations can easily be substituted by others present in solution. Barrer and MacLaeod [1] studied the enhancement of the adsorption capacity of smectites when exchanged with tetraalquilammonium salts. On this basis, and improving thermal stability the use of voluminous metallic-polioxocations was introduced by Brindley and Sempels [2] and Lahav et. al. [3]. Careful calcination of the exchanged clay leads to interesting materials with permanent microporosity across a range of molecular sizes. They are reported as PILCs (Pillared Inter-Layered Clays) because the calcined oxocations act as pillars that prop up clay layers. Since the work of Brindley and Sempels and Lahav et. al. different oxocations of Al, Zr, Ti, La, Ga, Fe and Cr have been used. Mixed pillars (Ga-Al, Ga-La) have been used to improve thermal stability. Several reviews on the topic have been reported [4-7]. PILCs are attractive as adsorbents or catalysts (either as catalysts or as catalyst supports). Their widespread use has been delayed by difficulties in obtaining a uniform and reproducible product at a pilot scale because of the many parameters [5] involved in the preparation process that influence PILCs’ properties. Several authors have extensively studied the preparation techniques for aluminum-pillared clays. Aouad et al. [8] have recently presented a novel method for Al-Pilc preparation. Our group has successfully prepared pillared clays [9, 10] with high specific surface area (360-389 m2/g) using different homoionic (Ca or Na) forms of the parent mineral clay. The process of converting the raw mineral to the homoionic form is time-consuming and expensive; therefore, the use of mineral with as little pretreatment as possible has been studied and the influence of the grinding of the starting montmorillonitic clay [11] reported. In this work, we study the influence of the starting material particle size in the textural properties of the pillared clays obtained using the mineral without any other pretreatment than drying, grinding and sieving. Thermal stability of the samples at 750oC is also reported. Experimental Clay from Bañado de Medina, Uruguay, was used. Geological, mineralogical and economic data for the deposit were first reported by Bossi [12]. Ford and Sergio [13] described the mineral as a calcium rich montmorillonitic clay, presenting a small degree of potassium substitution localized in some segregated layers. Studies of clay properties along the mine profile showed a slight increase (10%) of the Cation Exchange Capacity (CEC). Mössbauer spectroscopy revealed the presence of Fe3+, 1.8% of the total as Fe(IV). The remaining Fe(VI) is present in two different symmetries, 96.5% of it in a higher one. Details of this determination are not presented in this paper. The color of the mineral is light pink and presents black inclusions that can clearly be distinguished along the mine profile. Emission spectrochemical analysis of these inclusions showed high manganese content. Chemical analysis of the clay mineral is presented in Table 1. Mineral Pretreatment The mineral was selected from the level F of the deposit as described previously by Ford and Sergio [13]. After oven drying at 105ºC for 24 hours the mineral was ground in a Ketshmühle (WRb 90 lb/4p) mill. The particle size of this sample was under 2000 μm, and only 20% of it is over 1000 mm. Three different fractions were selected by sieving: under 250 mm (<250),>μm and 450 μm (250-450), and above 450 μm (>450), representing respectively 38.8%, 15.3% and 46.0% of the total mass of the dry mineral. Pillaring Agent The pillaring agent used was an aluminum polioxocation prepared by basic hydrolysis of an aluminum salt. A 0.2 M NaOH (Carlo Erba 98%) solution was poured at 3-4 ml/min over a 0.2M AlCl3 (Merck, anhydrous 97%) solution while stirred vigorously, until a molar ratio OH / Al = 2 was reached. The final pH was 3.5, and the solution was kept for 1 hour at 50ºC. The polioxocation crystal structure was determined by Johansson [14] to have a central aluminum in tetrahedral coordination surrounded by 12 aluminums in octahedral coordination. The cation formula is {AlO4Al12(OH)24(OH2)12}7+, and it is generally referred to as Al13. It is known that this structure remains unchanged in solution. According to Bottero et. al. [15], in the present preparation conditions, 90% of the Aluminum is embodied in the Al13 polication. Pillaring Ten grams of the mineral sample were suspended in deionized water (10% in weight) and stirred continuously for 16 hours. A suspending agent (Kelzan KC, Merck & Co, Inc. Kelco Division) was added (2% in weight) and stirring maintained for another 30 minutes. Afterwards the pillaring agent solution was added (4 ml/min) over the clay suspension while stirred vigourosly. The final ratio Aluminum /clay was 5 mmol per gram. The resulting suspension was kept at 80ºC for 3 hours and at room temperature for 16 hours. The solid was recovered by vacuum filtration and then repeatedly washed with deionized water at 80ºC until the wash water conductivity was less than 20 µS/cm. After the solid was oven dried at 60ºC the sample was calcined in a tubular furnace (Carbolite CTF-12/75) with programmable temperature controller (Eurotherm) at 1ºC/min up to 550ºC, and then kept at 550ºC for 2 hours. It is known that during the dehydration process the ionic bond between the clay lamella and the exchanged polication shifts to covalent in the PILC [3]. The samples are labeled as Mx where x represents the different particle size fraction as previously referred to. The pillared sample obtained after the milled whole mineral, without any fraction selection is labeled as M. Another M>450 PILC was prepared as previously described, but after aging at 80oC for 3 hours it was kept for 90 hours at room temperature before washing. This sample is labeled as M>450,aged. Thermal stability of the samples was controlled as follows: the Mx samples were calcined at 2oC/min up to 550oC and then at 1oC/min up to 750oC; the final temperature was maintained for 2 hours. Samples are labeled as Mx-750. Characterization Samples were characterized by X-Ray Diffraction (XRD), Differential Thermal Analysis (DTA), Cation Exchange Capacity (CEC) and Nitrogen adsorption isotherms. Table 1. Chemical Analysis of the mineral expressed as weight %. | SiO2 | 65.42 | MnO | 0.193 | Na2O | 0.13 | P2O5 | 0.02 | | Al2O3 | 12.10 | MgO | 3.31 | K2O | 0.17 | | | | Fe2O3 | 1.48 | CaO | 1.76 | TiO2 | 0.072 | LOI | 14.02 | LOI: Lost by ignition. Table 2. p/po range for the best fit of adsorption data of PILCs to different models used for textural characterization. | | | | | | | p/po | 7x10-3 – 0.1 | 2x10-3 – 5x10-2 | 0.80 – 0.95 | 0.01 – 0.1 | X-Ray Diffraction 1 g of the solid in 25 mL of sodium hexametaphosphatum solution (4.5 g/L) was maintained in an ultrasonic bath for 15 minutes. This suspension was diluted to 50 ml with deionized water and allowed to settle down in order to separate the particle size over 2 μm. 1-2 ml of the supernatant was spread over a glass slide and left to dry. Difractograms were obtained with a Phillips 1140 equipment, using CuKa radiation and operated at 40kV, 30 mA and 1º 2q /min. The d001 spacings (in nm) were determined using Bragg’s law. DTA Analysis A Shimadzu DTA-50 was used. A 15 mg sample was weighed in a Platinum capsule and heated up to 1100ºC at a rate of 10ºC/min in air. Cation Exchange Capacity Cation exchange capacity was determined by the ammonium acetate (1 M and pH = 7) method. Samples of 250 mg were exchanged with this solution three times and then washed with deionized water; a final wash was performed with anhydrous methanol. The amount of ammonium retained was determined using a distillation unit Kjeldtec 1002. The CEC is expressed as mili-equivalent per gram of the dry sample. For the raw mineral, the exchanged ammonium acetate solutions and washing water were recovered, diluted to an exact volume and used to determine the exchangeable cations concentrations: Ca2+ and Mg2+ using Atomic Absorption Spectroscopy and Na+ and K+ by Atomic Emission Spectroscopy. Nitrogen Adsorption-Desorption Isotherms Nitrogen adsorption-desorption isotherms were determined at -196oC in an Autosorb-1 (Quantachrome). Samples of 60 mg were outgassed at 370ºC under vacuum (final pressure less than 10-4 mbar). Textural parameters were derived from adsorption data. The specific surface area (s.s.a.) was determined based on BET [16] and Langmuir [17] models. The specific total pore volume (VT) was determined from the adsorption at the relative pressure of 0.95, converted to liquid volume assuming a nitrogen density of 0.808 g /mL. The specific micropore volume (VDR) was determined using the Dubinin-Radushkevich model [18]. The t-plot method [18] was used to determine the external specific surface area (s.s.a.ext) and the specific micropore volume (Vt mpore) using the t value given by Halsey equation [19]. The meso-macropore size distribution was analyzed based on the Barrer, Joyner and Halenda (BJH) model [20] and for micropore size distribution the methods proposed by Horvăth and Kawazoe (HK) [21] and by Saito and Foley (SF) [22] were used considering the oxidic character of the surface. The range of best fitting for each of the models is listed in Table 2. Results and Discussion PILC yield, in all cases, exceeded 90% of the dry mineral weight. The CEC for the raw mineral was 1.13 meq/g, and the composition in meq /g of dry sample: Ca2+ : 0.83; Mg2+ : 0.29; Na+ : 0.012 and K+ : 0.002. DTA Results Figure 1 shows the DTA profile for the mineral. The presence of two endothermic peaks at 115ºC and 200ºC can be assigned to the release of water molecules coordinated to calcium in the interlayer. The peak at 648ºC is related to the release of OH groups [23] from the Gibbsite octahedral layer. The exothermic peak at 900oC corresponds to the collapse of the clay structure [24]. | 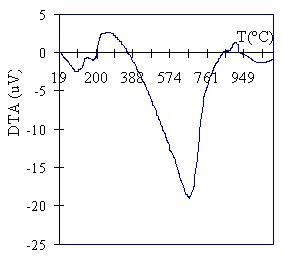
| | Figure 1. DTA for the mineral. | X-Ray Diffraction Results Figure 2 shows X-ray Diffractograms compared with the axis as indicated. Peaks corresponding to the d001 spacing are shown. For clarity, only the M<250 diagram is presented: all the others superposed this one. The d001 peak for the mineral corresponds to a 1.47 nm spacing that is characteristic for a calcium montmorillonite. This observation coincides with the exchange cation composition. The peak for the pillared samples corresponds to a d001 spacing of 1.80 nm, and it is indicative of the efficiency of the pillaring process in expanding the basal spacing and producing the microporosity of the sample. The relative intensity of the peaks suggests a crystallinity difference between the mineral and the PILC, but even at lower intensity, the diffraction peak for the PILC is well defined. Supposing a layer thickness of 0.96-0.98 nm, an interlayer free space of 0.83 nm can be deduced for the PILC. This value corresponds to the height of the pore gallery. | 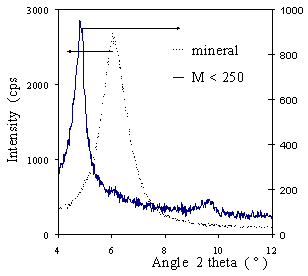
| | Figure 2. DRX diagram for the mineral and sample M<250. | Nitrogen Adsorption-Desorption Isotherms Nitrogen adsorption-desorption isotherms are shown in Figure 3. The filled symbols represent adsorption, while the open ones represent desorption data for the pillared samples; the isotherm for the starting raw mineral is drawn as a line. | 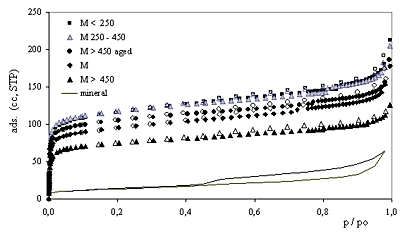
| | Figure 3. Nitrogen adsorption-desorption isotherms for samples calcined at 550oC: see explanation in the text. | The isotherms for the samples M<250 and M250-450 nearly coincide, though that for M250-450 is represented by grey symbols in order to facilitate its identification. Pillaring increases the adsorbed amount of nitrogen at low relative pressures but does not alter the intermediate zone of the isotherms (all run parallel to each other). The PILCs’ isotherms present a steep adsorption at very low relative pressures characteristic of microporous solids. All the isotherms are type IV isotherms of the BDDT classification [25] and present a small hysteresis loop type H4 as defined by IUPAC [26], characteristic of the presence of mesopores between parallel plates particles. Samples M<250 and M250-450 (prepared using the smaller mineral particle size) present the higher adsorption and sample M>450 (prepared using the coarser starting clay) presents the lowest adsorption of the series in the low-pressure region. The pillared mineral (M) isotherm lies between the M>450 and the M<250>isotherms. Pillaring increases the amount adsorbed at low relative pressures. The small difference of adsorption between the branches of the hysteresis loop suggests the small contribution of mesopores to the amount adsorbed. The parallel branches at intermediate relative pressures amongst all the isotherms (including that for the mineral) suggests that the pillaring process does not alter the layered structure of the clay nor the external specific surface area of the parent mineral. Textural Parameters for PILCs Calcined at 550oC Textural parameters for all the samples derived from nitrogen adsorption isotherms data are shown in Table 3. Table 3. Textural parameters for PILCs calcined at 550oC. | | | | | | | | | | | | | | | | | M | 381 | 355 | 0.212 | 0.147 | 9.4 | 0.67 | 0,43 | 27 | 0.154 | | M<250 | 463 | 431 | 0.256 | 0.179 | 8.9 | 0.72 | 0,43 | 37 | 0.170 | | M250-450 | 475 | 441 | 0.252 | 0.182 | 9.6 | 0.69 | 0,43 | 34 | 0.169 | | M >450 | 302 | 280 | 0.163 | 0.117 | 9.2 | 0.69 | 0,43 | 27 | 0.114 | | M>450, aged | 425 | 394 | 0.222 | 0.164 | 9.2 | 0.75 | 0.45 | 29 | 0.174 | Micropore diameters (dmpore) determined by the SF and HK models are presented. These two models were deduced on different bases. The HK model was originally applied to nitrogen adsorption in the slit shaped pores of activated carbon. The SF model was proposed to accommodate the HK method to the cylindrical pore network of zeolites, using the same potential energy of interaction and considering the oxidic character of the surface. The results we present in this paper were derived using the same interaction parameter in both calculations. The physical properties of nitrogen and oxygen involved were taken respectively from references [21] and [22]. Under these conditions, the pore openings derived from the XRD data are twice those derived using the HK model, but differ only in 0.1nm from those determined by the SF model. The fact could be explained if even when a slit-like pore network is expected for PILCs because of the layered clay structure, a cylindrical-like shaped pore network could result if pillars were distributed in a relatively high and uniform density on the host clay lamella. As seen in Table 3 the s.s.a. of the samples is related to the mineral particle size selected for pillaring. The value derived using Langmuir model is, in every case, about 7% higher than that obtained by the BET plot. There is no significant difference between the s.s.a. for samples M<250>and M250-450, 431 and 441 m2/g respectively, the higher of the series. The lower s.s.a. corresponds to sample M>450 prepared using the coarser particles, 35% lower than that for the finer fractions. It is also worth noting that the s.s.a. for M sample is very close to the weighed average value determined using the particle size distribution. As the Langmuir and BET models are not based on the same assumptions, we would not expect to find a coincidence between the values determined by each of these models. Indeed, for microporous solids an overestimation of the specific surface area derived from either of these models is expected. In fact, even when not every nitrogen molecule adsorbed in the micropores is in contact with the solid surface, they are all assigned to s.s.a. calculation resulting in the mentioned overestimation. A quite good agreement between both values in this case can be explained by the value of the pore gallery height. As determined based on XRD, the d001 spacing of 0.8 nm is just enough to accommodate inside a micropore only two monolayers of adsorbed Nitrogen molecules, with a diameter of 0.35 nm. In this way both molecules are being assigned to a real surface, which is not the case for wider pore diameters that can accommodate more than two molecules [9, 27]. For all the samples the specific micropore volume represents approximately 70% of the specific total pore volume of 0.250 mL/g (which includes micro, meso and macropores). The difference between these two values is attributed to the specific mesopore volume. This coincides with the small hysteresis loops of all the isotherms. When the BJH method was used to analyze mesopore size distributions, no peak was found for any of the studied samples, suggesting a wide mesopore size distribution, attributable to interparticle pores. The specific micropore volumes determined after DR and t-plot diagrams show a high coincidence. Thermal Stability Figure 4 shows the thermal stability of the samples at 750oC determined from nitrogen adsorption isotherms. Isotherms for samples Mx and Mx-750 are shown in the same diagram for comparison. The isotherms are parallel for all the samples, enforcing the idea that the layered structure and the external s.s.a. are not affected by calcination at 750oC. The isotherms for M>450,aged coincide showing a very strongly thermal stability for this sample. | 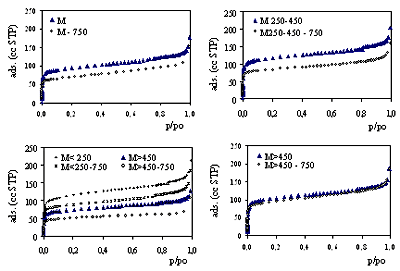
| | Figure 4. Thermal stability at 750oC. Nitrogen adsorption isotherms at 550oC and 750oC are shown for each sample as indicated in the legends. | Textural Parameters for PILCs Calcined at 550oC Textural parameters for the Mx-750 samples derived from nitrogen adsorption isotherms data are shown in Table 4. Calcination at 750oC produces a slight decrease of the micropore diameter, from 0.7 nm for the Mx samples to 0.64 nm for the Mx-750 samples, determined by either the HK or SF methods. With the exception of M>450,aged, calcination at 750oC diminished the s.s.a. (in about 100 m2/g) and VTotal and Vmpore (in about 30-60 μL/g). Table 4. Thermal stability at 750oC. Textural parameters for samples Mx-750 derived from nitrogen adsorption isotherms. | | | | | | | | | | | | | | | | | M | 278 | 259 | 0.169 | 0.107 | 9.7 | 0.67 | 0.39 | 32 | 0.116 | | M<250 | 354 | 330 | 0.206 | 0.155 | 7.5 | 0.64 | 0.39 | 37 | 0.146 | | M250-450 | 337 | 313 | 0.184 | 0.128 | 10.6 | 0.64 | 0.39 | 33 | 0.130 | | M >450 | 200 | 187 | 0.104 | 0.077 | 9.6 | 0.64 | 0.40 | 17 | 0.077 | | M>450, aged | 377 | 352 | 0.219 | 0.144 | 10.4 | 0.64 | 0.39 | 41 | 0.151 | In order to evaluate thermal stability we introduce a thermal stability parameter (TSP) defined as TSP = 100* (value750/ value550). TSP simplifies the comparison of the textural parameter values shown in Table 3 and Table 4 and quantifies the changes in isotherms shown in Figure 4 to evaluate thermal stability. Its values for Langmuir and BET s.s.a., VTotal , VDR and Vt-mpore are shown in Table 5. Comparing the TSP values for samples prepared with 16 hours aging (all except the last one listed in the table) it is interesting to note that even when the absolute difference between micropore adsorption at 550oC and 750oC is high, specially for samples M250-450 and M<250, the TSP value does not significantly differ from that for the M sample. Table 5. Thermal stability parameter (TSP) as defined in the text. | | | | | | | | | | M | 73 | 73 | 80 | 73 | 75 | | M<250 | 76 | 77 | 80 | 87 | 86 | | M 250-450 | 71 | 71 | 73 | 70 | 77 | | M>450 | 66 | 67 | 64 | 66 | 68 | | M>450 aged | 89 | 89 | 99 | 88 | 87 | M>450 displays poorer thermal stability evaluated as a TSP value. M<250 displays higher stability of the series. Though the stability of textural parameters, when the PILC is heated at 750oC, is related to the particle size of the starting clay selected for pillaring. In fact, the coarser grinded mineral gives the lowest thermal stability and the finer particle size produces more stable structures when heated to 750oC. The best thermal stability is attained by sample M>450,aged. This sample was obtained by pillaring the coarser starting particle size but it was aged for 90 hours instead of 16 hours after exchange with the Al13 polication and before washing. This procedure seems to have contributed greatly to the thermal stability of PILC. Its influence can be explained by the fact that a more uniform distribution of the pillars in the interlayer space is facilitated by the longer aging period. During the exchange process and in the first step, the oxocations present in solution are crowded at the edges of the clay particles; during the aging process the cations migrate in the interlamellar space, resulting in a more uniform density on the lamella area. Figueras [5] has postulated the homogeneity of the pillar density in the interlamellar space as responsible for the thermal stability of PILCs. Conclusions A preparing method for PILCs that does not require any other mineral pretreatment than drying and grinding is presented. The preparation technique allows a high mineral yield and produces interesting microporous solids with well-defined structure, high specific surface area and good thermal stability at 750oC. Micropore size distributions are monomodals, and pore openings are not dependent on the particle size of the mineral selected for pillaring. Micropore diameter determined based on the SF model is coincident (difference of 0.1nm) with the pore gallery height determined based on XRD diffractograms. This fact suggests that the pore network more closely resembles that of zeolites than the slit-shaped one of carbons, though the distance height of the pore gallery should be of the same order. The PILC obtained from the coarse particle size of the mineral presents the lower values of s.s.a. and pore volume and the lowest thermal stability. The highest s.s.a. and pore volume are obtained when the finer particle sizes of the parent clay are pillared. For particles sizes below 450mm no difference of the textural parameters here evaluated was observed. When the aging period following the polication exchange for the coarser fraction is increased to 90 hours the resulting PILC thermal stability reaches values of 90%, as noted in this paper. A high mineral yield and a simpler manipulation can yield economic advantages in producing microporous solids with high mineral yield to be used in catalysis and adsorption processes, especially if size or form selectivity are required. |
<< Home